![]()
Rparadox provides a simple and efficient way to read
data from Paradox database files (.db) directly into R as
modern tibble data frames. It uses the underlying
pxlib C library to handle the low-level file format details
and provides a clean, user-friendly R interface.
This package is designed to “just work” for the most common use case:
extracting the full dataset from a Paradox table, including its
associated BLOB/memo file (.mb).
.db
files without needing database drivers or external software.tibble format, which is fully compatible with the Tidyverse
ecosystem..mb)
files.Date,
Time (hms), Timestamp
(POSIXct), Logical, Integer,
Numeric, and binary blob objects.# stable version from CRAN
install.packages("Rparadox")You can install the development version of Rparadox from GitHub using
the devtools package.
# install.packages("devtools")
devtools::install_github("celebithil/Rparadox")read_paradox() functionThe easiest way to read a Paradox file is with the high-level
read_paradox() function. It handles opening the file,
reading the data, and closing the connection in a single step.
# 1. Load the package
library(Rparadox)
# 2. Get the path to an example database
db_path <- system.file("extdata", "biolife.db", package = "Rparadox")
# 3. Read the data directly into a tibble
# This automatically finds 'biolife.mb' and handles data types.
biolife_data <- read_paradox(db_path)
# 4. View the data
print(biolife_data)
#> # A tibble: 28 × 8
#> `Species No` Category Common_Name `Species Name` `Length (cm)` Length_In
#> <dbl> <chr> <chr> <chr> <dbl> <dbl>
#> 1 90020 Triggerfish Clown Trig… Ballistoides … 50 19.7
#> 2 90030 Snapper Red Emperor Lutjanus sebae 60 23.6
#> 3 90050 Wrasse Giant Maor… Cheilinus und… 229 90.2
#> 4 90070 Angelfish Blue Angel… Pomacanthus n… 30 11.8
#> 5 90080 Cod Lunartail … Variola louti 80 31.5
#> 6 90090 Scorpionfish Firefish Pterois volit… 38 15.0
#> 7 90100 Butterflyfish Ornate But… Chaetodon Orn… 19 7.48
#> 8 90110 Shark Swell Shark Cephaloscylli… 102 40.2
#> 9 90120 Ray Bat Ray Myliobatis ca… 56 22.0
#> 10 90130 Eel California… Gymnothorax m… 150 59.1
#> # ℹ 18 more rows
#> # ℹ 2 more variables: Notes <chr>, Graphic <blob>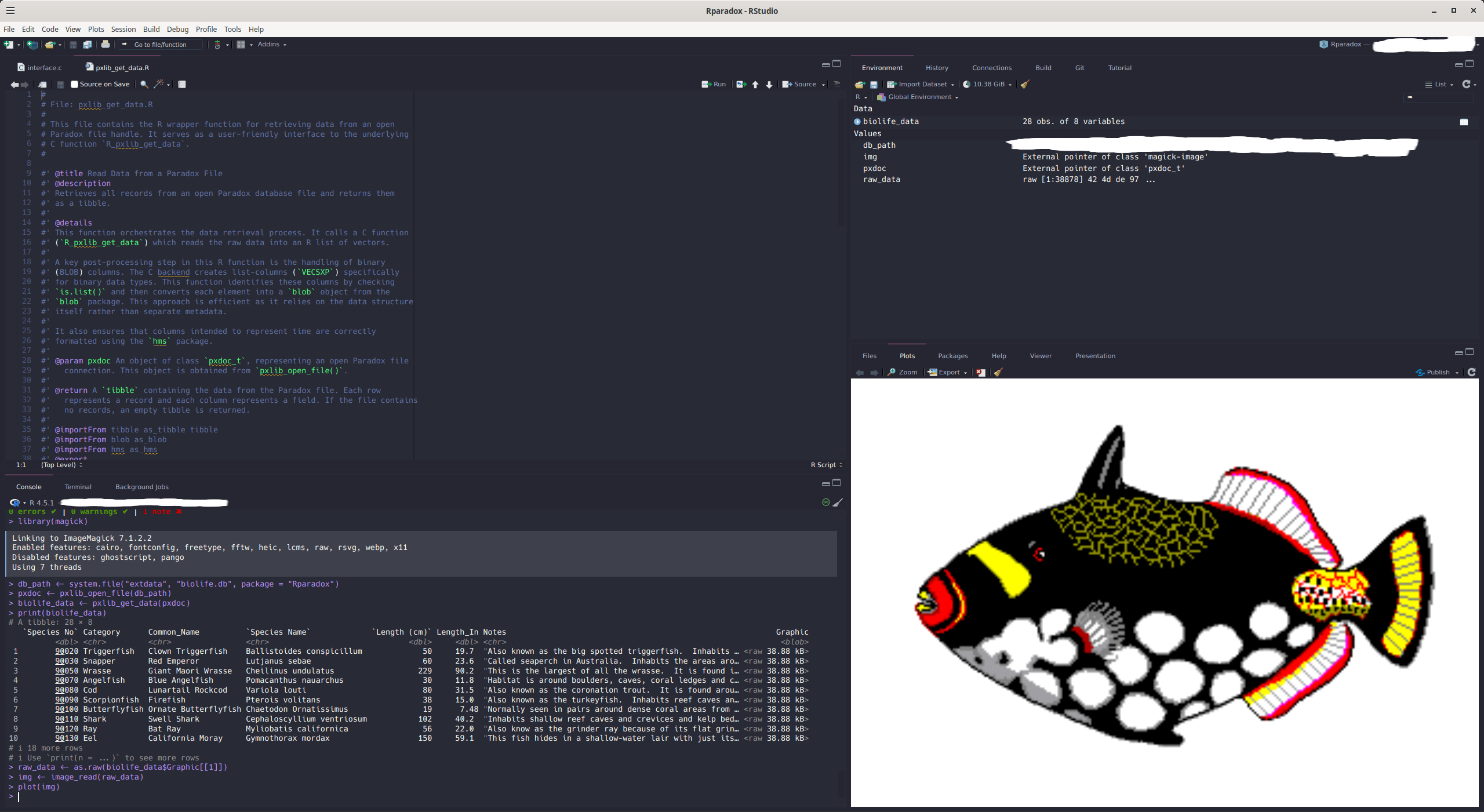
If you have a legacy file where the encoding is specified incorrectly
in the header, you can manually override it using the
encoding parameter with read_paradox().
library(Rparadox)
# This tells the package to interpret the source data as CP866
data <- read_paradox("path/to/your/file.db", encoding = "cp866")This ensures that all text fields are correctly converted to UTF-8 in
the final tibble.
For more control, you can use the lower-level functions. This is useful if you want to inspect metadata before reading the full dataset.
library(Rparadox)
db_path <- system.file("extdata", "biolife.db", package = "Rparadox")
# 1. Open the file and get a handle
pxdoc <- pxlib_open_file(db_path)
if (!is.null(pxdoc)) {
# 2. Get metadata without reading all the data
metadata <- pxlib_metadata(pxdoc)
cat("Number of records:", metadata$num_records, "\n")
# 3. Read the actual data
data_table <- pxlib_get_data(pxdoc)
# 4. Always close the file when you're done
pxlib_close_file(pxdoc)
}
#> Number of records: 28